
厚生労働省は、「臨床研究において使用される未承認の医薬品、医療機器及び再生医療等製品の提供等に係る医薬品、医療機器等の品質、有効性及び安全性の確保等に関する法律の適用について」注1)において、未承認医療機器の提供等に係る薬機法適用の基本的な考え方を示しています。この中で、企業が未承認医療機器を提供して実施する場合の臨床研究の要件として、医師又は歯科医師が主体的に実施する臨床研究であることや、企業等が医師又は歯科医師の求めに応じて未承認医薬品等及びこれらに関する必要な情報を提供等することを挙げています。また、「医師又は歯科医師が責任主体となっていない場合、臨床研究を目的とする一連の提供行為の正当性を担保することが困難となること」などが記載されています。また、留意事項には、機器提供者と臨床研究を実施する医師又は歯科医師に対する遵守事項も記載されていますので、未承認医療機器を用いた臨床研究を実施する場合はご参照下さい。
この通知の発出に伴い、「臨床研究において用いられる未承認医療機器の提供等に係る薬事法の適用について」(2010 年3月31 日 薬食発0331 第7号厚生労働省医薬食品局長通知)と「「臨床研究において用いられる未承認医療機器の提供等に係る薬事法の適用について」に関する質疑応答集(Q&A)について」(2011年3月31日薬食監麻発0331第7号)は廃止されました。
なお、製造・販売の「承認」、「認証」、又は「届出」が行われていない医療機器は全て「未承認医療機器」になります。また、承認等が得られた医療機器であっても、承認等の範囲外で形状・仕様等を変更した場合や承認等の範囲外の目的で使用する場合も未承認医療機器と同じ扱いになります。また、未承認医療機器の場合、「通常の診療を超える医療行為」であって、研究目的で実施するものは、侵襲の程度にかかわらず全て、「介入」注2)に分類されることに留意ください。
(2020年1月24日時点の記載です。)
(注1)厚生労働省 医薬・生活衛生局長通知薬生発0406 第3号「臨床研究において使用される未承認の医薬品、医療機器及び再生医療等製品の提供等に係る医薬品、医療機器等の品質、有効性及び安全性の確保等に関する法律の適用について」(平成30年4月6日)
(注2)介入とは、「研究目的で、人の健康に関する様々な事象に影響を与える要因(健康の保持増進につながる行動及び医療における傷病の予防、診断又は治療のための投薬、検査等を含む。)の有無又は程度を制御する行為(通常の診療を超える医療行為であって、研究目的で実施するものを含む。)」を指します。(「人を対象とする医学系研究に関する倫理指針」から)
「人を対象とする医学系研究に関する倫理指針ガイダンス 」には、「「リスク」とは、研究の実施に伴って、実際に生じるか否かが不確定な危害の可能性を指す。その危害としては、身体的・精神的な危害のほか、研究が実施されたために被るおそれがある経済的・社会的な危害が考えられる。」と、臨床研究を実施する上でのリスクが記載されています。それでは、医薬品、医療機器それ自体が持つリスクとは何でしょうか?医薬品は副作用を中心としたリスクの考え方があります。これに対して、医療機器は不具合の結果による健康被害でリスクが判断されます。
医療機器の国際分類は、”不具合が生じた場合に”、人にどの程度の健康被害を与えるかによって、下図のようにクラスⅠからⅣのリスクに応じた分類になっています。クラスⅠは人体へのリスクが極めて低いもの、クラスⅡは人体へのリスクが比較的低いもの、クラスⅢは人体へのリスクが比較的高いもの、クラスⅣは生命の危機に直結する高いリスクがあるものになります。
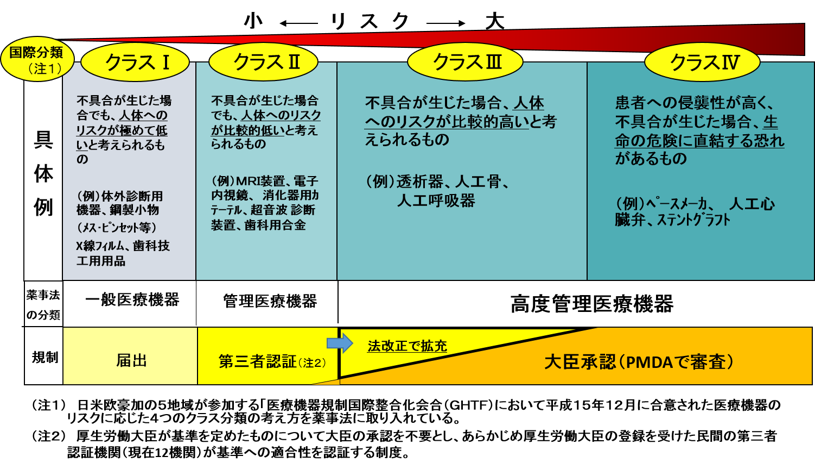
独立行政法人 医薬品医療機器総合機構 ホームページの資料より
それでは、「未承認」の医療機器は高リスク機器、「非侵襲」の医療器機器は低リスク機器でしょうか? 必ずしもそうではありません。未承認であっても、リスクの極めて低いと考えられる医療機器もあります。非侵襲の検査機器であっても診断結果が治療に大きな影響を及ぼす場合は、高リスク機器になります。医療機器自体が持つリスクは、承認・未承認の違いや侵襲注1)の程度と必ずしも一致しないことに注意してください。
医療機器のクラス分類は機器に対する薬機法の規制による分類ですが、臨床研究では、「人を対象とする医学系研究に関する倫理指針ガイダンス 」に示されているように、医療機器自体のリスクだけでなく、臨床研究のプロトコルの中でどのように使用されるかによってもリスクが変わることに留意する必要があります。また、医療機器の人体へのリスクは、患者さんだけでなく、操作者(医療者)に対しても考慮しなければなりません。
(2020年1月24日時点の記載です。)
(注1)侵襲とは、「研究目的で行われる、穿刺、切開、薬物投与、放射線照射、心的外傷に触れる質問等によって、研究対象者の身体又は精神に傷害又は負担が生じること」を言います。 また「侵襲のうち、研究対象者の身体及び精神に生じる傷害及び負担が小さいものを「軽微な侵襲」」と言います。(「人を対象とする医学系研究に関する倫理指針 」から)
医療機器の製造・販売において、臨床的な有効性及び安全性が性能試験、動物試験等の非臨床試験成績又は既存の文献等のみによって評価できる場合は、治験成績が不要注1)となります。したがって、機器の基本的な性能や安全性を担保するための非臨床試験が非常に重要になります。具体的には、医療機器の承認を得るにあたっては、医療機器の設計において、物理的、化学的、生物学的、電気的安全性等が要求され、医用電気機器に対する基本的要求事項として、国際規格(IEC60601-1)や生物学的安全性の規格(ISO10993-1)などが各国の国内規格として取り入れられています。さらに、医用電気機器では、電磁両立性に関する規格(IEC60601-1-2)により、妨害電磁波に対する耐性や電磁波放射ノイズが規制され、同規格に適合することが要求されています。
臨床研究を実施する場合も、機器の基本的な性能や安全性を担保するための非臨床試験の結果が重要であることに変わりはありません。特に、未承認医療機器を使用する場合は、「臨床研究機器に関する説明書テンプレート」などを参考に、機器提供企業から電気安全性、生物学的安全性などの安全性試験や性能評価に関する試験結果を入手する必要があります。
なお、医療機器の場合、操作方法の習得や保守管理が、研究対象者(患者等)のみならず操作者(医療者)の安全性の観点から重要になります。必要に応じて、操作トレーニング実施計画等を立案・作成することが望まれます。また、医療機器の保管・管理の体制整備も重要です。具体的には、医療機器提供企業と事前に相談し、医療機関側の保管場所、保管責任者、保守メンテナンスなどの保管プロセスを確立するとともに、保管・管理の書類整備を行う必要があります。
(2020年1月24日時点の記載です。)
(注1)「医療機器の臨床的な有効性及び安全性が性能試験、動物試験等の非臨床試験成績又は既存の文献等のみによっては評価できない場合に、臨床試験の実施が必要となり、臨床試験成績に関する資料の提出」が求められます。ここで臨床試験は治験を指しています。ただし、「臨床試験の試験成績に関する資料の要否については、個々の医療機器の特性、既存の医療機器との同等性、非臨床試験の試験成績等により総合的に判断」されます。(「医療機器に関する臨床試験データの必要な範囲等について」(平成20年8月4日薬食機発第0804001号通知)から)
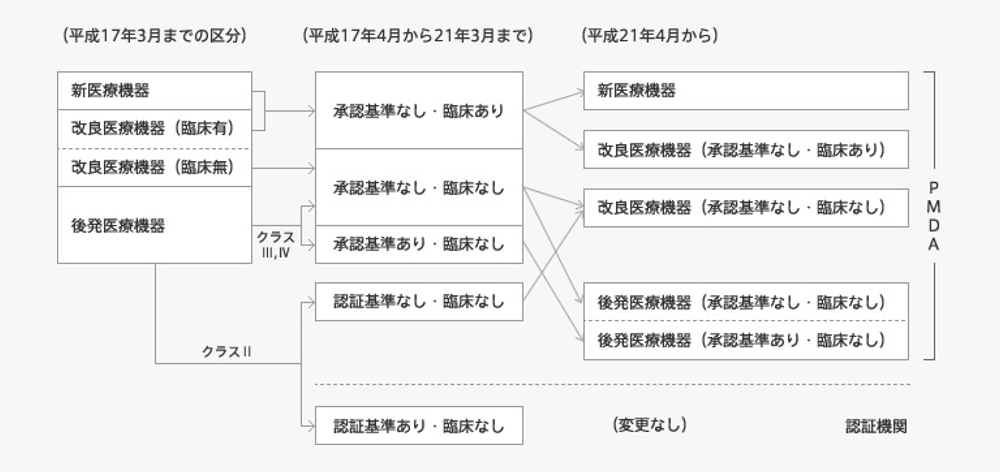
医療機器の製造・販売には、「届出」、「認証」あるいは「承認」が必要になります。リスクが極めて低いクラスⅠの医療機器は医薬品医療機器総合機構(PMDA)への届出(自己認証)のみになり、臨床試験成績(治験成績)の提出は不要です。また、大部分のクラスⅡ、一部のクラスⅢの医療機器には認証基準注1)があり、第三者認証機関が審査します。この場合も臨床試験成績の提出は不要です。このため、治験の対象は、PMDAによる承認審査を受ける一部のクラスⅡやクラスⅢ、Ⅳの医療機器に限られます。
PMDAによる承認審査は、医療機器は改良・改善で進化するものが多いため、下図のように新医療機器、改良医療機器、後発医療機器の3つのトラック注2)に分かれ、また、臨床試験成績の要否(「臨床あり」、「臨床なし」)、承認基準の有無に基づいて行われます。ここで、原則、治験が必要なもの(「臨床あり」)は、さらに、新医療機器や改良医療機器の一部に限定されます。
PMDAで承認された医療機器の品目数を、承認申請区分別で見ると分かりますが、臨床試験成績を必要としない改良医療機器(臨床なし)や後発医療機器(臨床なし)の件数が多く、全体の90%を超えています。
承認申請区分別 医療機器承認品目数
| 平成26年度 | 平成27年度 | 平成28年度 | 平成29年度 | 平成30年度 | |
| 新医療機器 | 67 | 56 | 26 | 27 | 38 |
| 改良医療機器(臨床あり) | 35 | 53 | 44 | 42 | 52 |
| 改良医療機器(臨床なし) | 213 | 240 | 225 | 215 | 216 |
| 後発医療機器 | 917 | 868 | 825 | 868 | 799 |
独立行政法人 医薬品医療機器総合機構 平成30事業年度業務実績より
また、全体の10%にも満たない臨床試験成績が提出された承認品目を見ると、ステント・カテーテルなどの海外製品のものが多数を占めるため、「海外の臨床試験成績のみ」や「臨床評価報告書注3)」で承認を受ける割合が多くなっています。
臨床試験成績が提出された承認品目数の内訳
| 平成26年度 | 平成27年度 | 平成28年度 | 平成29年度 | 平成30年度 | |
| 国内の臨床試験成績のみ | 10 | 23 | 9 | 14 | 14 |
| 海外の臨床試験成績のみ | 24 | 23 | 25 | 26 | 31 |
| 国際共同治験 | 0 | 2 | 3 | 2 | 1 |
| 臨床評価報告書 | 37 | 23 | 13 | 11 | 19 |
| その他 | 5 | 10 | 4 | 2 | 5 |
注)その他は、海外臨床試験成績+国内臨床試験成績を併用した品目など
独立行政法人 医薬品医療機器総合機構 平成30事業年度業務実績より
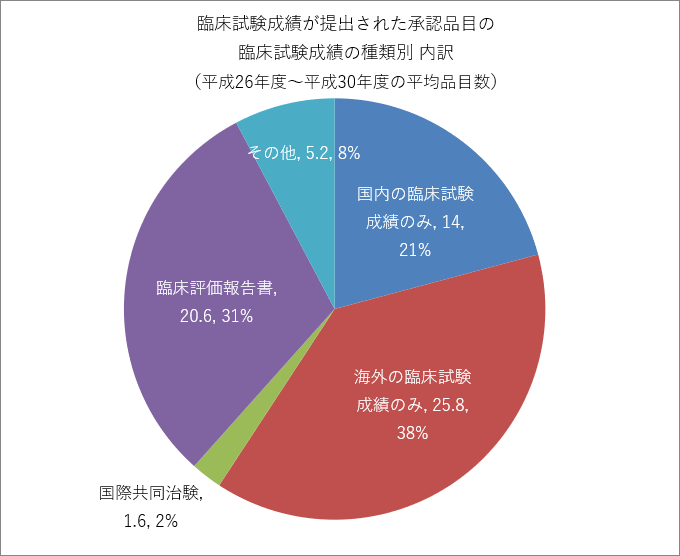
医療機器の場合、臨床試験成績を必要とする品目が非常に限られていること、さらに、臨床試験成績を必要とするリスクの高い医療機器は海外製品が多いため、国内での臨床試験(治験)を必ずしも要求していないことが、医薬品と大きく異なるところです。
一方、「届出」、「認証」、「承認」において臨床試験成績の提出が不要であっても、改良・改善で進化する医療機器では、臨床研究による評価が非常に重要になります。このため、医療機器の有効性・安全性の検証的な評価とともに、医療機器の性能評価やユーザビリティ注4)の評価などを含め、多くの探索的臨床研究が実施されています。
(2019年12月18日時点の記載です。)
(注1)厚生労働大臣が基準を定めたものについて大臣の承認を不要とし、あらかじめ厚生労働大臣の登録を受けた民間の第三者認証機関が基準への適合性を認証する制度で用いられる適用範囲、技術的な基準、使用目的又は効果で構成された医療機器の認証基準
(注2)新規性の程度によって審査プロセスを明確化するために導入された3区分(新医療機器・改良医療機器・後発医療機器)による審査体制
(注3)医療機器の臨床的な有効性及び安全性が性能試験、動物試験等の非臨床試験成績又は既存の文献等のみによっては評価できない場合は、臨床試験(治験)の実施が必要です。しかし、臨床試験成績に関する資料の提出が必要な範囲の機器であっても、既存の文献等によって評価可能と考えられる場合には、「臨床評価報告書」でもって臨床試験成績に代えることが出来ます。
(注4)医療機器のユーザビリティが適切でないために起こる使用時のエラーによるリスクを低減することが益々重要になっています。このため、2015 年に発行された IEC 62366-1を基に、国内においても、2019年10月1日にJIS規格(JIS T 62366-1:2019 医療機器―第1部:ユーザビリティエンジニアリングの医療機器への適用)が制定されました。また、同年10月3日には厚生労働省から「ユーザビリティエンジニアリングの医療機器への適用に関する日本産業規格の制定に伴う医薬品、医療機器等の品質、有効性及び安全性の確保等に関する法律上の取扱いについて」(薬生発1001第1号・薬生監麻発1001第5号)が発出されています。
Q4に示したように、「医療機器の臨床的な有効性及び安全性が性能試験、動物試験等の非臨床試験成績又は既存の文献等のみによっては評価できない場合は、臨床試験(治験)の実施が必要です。しかし、臨床試験成績に関する資料の提出が必要な範囲の機器であっても、既存の文献等によって評価可能と考えられる場合には、「臨床評価報告書」でもって臨床試験成績に代えることが出来ます。」注1)となっています。
しかし、「動物試験等の非臨床試験成績又は既存の文献等のみによっては評価できない場合」の解釈は明確ではありません。これに関して、厚生労働省は、2017年に「医療機器の「臨床試験の試験成績に関する資料」の提出が必要な範囲等に係る取扱い(市販前・市販後を通じた取組みを踏まえた対応)について」の通知注2)を発出しています。
これは、医療機器は改良、改善が頻繁かつ多様な内容で行われる等の特性があるため、それらの特性を活かしつつ、開発をより効率的に行う観点から検討されたもので、市販前から市販後まで一貫した安全性及び有効性の確保策を実施することにより、市販前の新たな治験実施の有無によらず、承認申請を行い得ると考えられるケースの取扱いを整理したもの注1)となっており、リバランス通知と呼ばれています。
この通知では、以下の3つケースについて具体的な対応が示されています。
- 国内医療環境への適合性を評価するための治験の取扱い
- 追加的な臨床的付加価値が比較的小さく、重大なリスクが想定されない改良医療機器の治験の取扱い
- 診断の参考情報となり得る生理学的パラメータを測定する診断機器に関する相談
1は外国で実施された治験成績の外挿性、2は低リスクの改良医療機器(臨床あり)に相当する医療機器、3は診断支援機器に関するものです。
3では、これまで「治験」が求められていた「新しい診断指標」の数値等に関して、最終目標の臨床的意義が確立されていない段階において、「使用目的又は効果」の範囲を限定することで承認申請を行うための道筋が示されています。これには、添付文書に「測定データはあくまで定性的であり、ひとつの参考情報であるため、他の情報とともに医師が総合判断すること」や「参考情報であり、診断の妥当性を保証するものではない。」などの制約が付きますが、承認後、臨床的エビデンスを確立した段階で一部変更申請などを行い、最終目標である臨床的意義を標榜することが出来ます。なお、この通知では、対象は、診断の参考情報を提供する低リスクのモニタリング医療機器となっています。
一定程度の臨床データが入手可能な、医療上の必要性の高い医療機器については、2017年に、使用条件の設定、市販後のデータ収集などの製造販売後のリスク管理を適切に行う事を前提に、限られた臨床データを基に承認申請を可能とする「革新的医療機器条件付早期承認制度」が実施されています。また、2020年9月1日に施行された薬機法の一部を改正する法律において、「医療機器等条件付き承認制度」注3)として法制化されました。
この承認制度では、2つの類型が対象となっています。
類型1は、以下の要件の全てに該当する医療機器又は体外診断用医薬品を対象としており、製造販売後リスク管理措置を適切に実施することを条件に、新たな治験を実施することなく、当該医療機器の安全性、有効性等を確認し、承認することになっています。
(類型1)
- ア.生命に重大な影響がある疾患又は病気の進行が不可逆的で日常生活に著しい影響を及ぼす疾患を対象とすること。
- イ.既存の治療法、予防法若しくは診断法がないこと、又は既存の治療法等と比較して著しく高い有効性又は安全性が期待されること。
- ウ.一定の評価を行うための適切な臨床データを提示できること。
- エ.新たな臨床試験又は臨床性能試験の実施に相当の困難があることを合理的に説明できること。
- オ.関連学会と緊密な連携の下で、適正使用基準を作成することができ、また、市販後のデータ収集及びその評価の計画を具体的に提示できること。
また、類型2は、以下の要件の全てに該当する、焼灼、遮蔽等の身体に物理的影響を与えることを目的とした医療機器又は体外診断用医薬品で、特定の疾病領域にかかる臨床データ等があり、他領域にも外挿可能と考えられるものが対象となっており、PHOENIX(Physical OpEratioN Items’ eXtrapolative and inclusive approval)制度と呼ばれています。
これは、手術ロボットでの反省を踏まえ、施設や術者等の限定や市販後安全対策の充実強化により、機器のもつ機能に着目した他臓器や部位への迅速な適用追加を実現するものになります。
(類型2)
- ア.焼灼その他の物的な機能により人体の構造又は機能に影響を与えることを目的とする医療機器又は体外診断用医薬品であって、医療上特にその必要性が高いと認められるものであること。
- イ.既存の臨床データでは直接的に評価されていない適用範囲に関する有効性及び安全性について、一定の外挿性をもって評価を行うための適切な臨床データを提示できること。
- ウ.新たな臨床試験又は臨床性能試験を実施しなくとも、その適正な使用を確保できることを合理的に説明できること。
- エ.関連学会と緊密な連携の下で、適正使用基準を作成することができ、また、市販後のデータ収集及びその評価の計画を具体的に提示できること。
医療機器は、低リスクから高リスク、小型から大型まで多種多様のものが存在し、市販後も継続的な改良・改善が繰り返されます。こうした医療機器の特性に応じた承認制度が順次施行されていますので、引き続き、今後の動向を注視する必要があります。
(2021年6月30日時点の記載です。)
(注1)薬食機発第0804001号通知「医療機器に関する臨床試験データの必要な範囲等について」(平成20年8月4日))
(注2)薬生機審発1117第1号・薬生安発1117第1号通知「医療機器の「臨床試験の試験成績に関する資料」の提出が必要な範囲等に係る取扱い(市販前・市販後を通じた取組みを踏まえた対応)について」(平成29年11月17日)
(注3)薬生機審発0831第2号通知「医療機器及び体外診断用医薬品の条件付き承認の取扱いについて」(令和2年8月31日)
2020年9月1日に薬機法の一部を改正する法律が施行され、医薬品、医療機器等をより安全・迅速・効率的に提供することを目的に、「先駆け審査指定制度注1)」や「条件付き早期承認制度注2)」(Q5 参照)、また、医療機器等の変更計画の確認及び計画に従った変更に係る「事前届出制度」が法制化されました。
これまでの一部変更申請では、市販後に恒常的に性能等が変化する人工知能関連技術を用いた医療機器や、市販後に収集するリアルワールドデータを利用して改良を行う医療機器、さらに、使用性向上のためのオプション部品等の追加では、継続的な改良を迅速に行うことは困難でした。
「医療機器の特性に応じた承認制度である事前届出制度」注3)は、改良が予め見込まれるものについては、「変更計画」を審査の中で事前に確認することで、計画された範囲内の承認事項の一部変更は、変更申請又は変更届出として、迅速に認める承認審査制度で、通称IDATEN: Improvement Design within Approval for Timely Evaluation and Noticeと呼ばれています。
IDATEN制度では、例えば、人工知能関連技術を用いた医療機器の場合、「変更計画確認申請書」注3,4)の添付資料として、審査の過程で、「変更計画の作成及び実施に関する手順」と、市販後に変化する性能を管理するための「その他人工知能関連技術の適正かつ円滑な管理に必要な資料」を提出し、変更計画に従った変更を実施することになります。その際、使用されることで性能等が変化しても、性能等の低下は許されないとの考え方から、製造販売業者等による管理が特に重要となっています。このため、性能等の向上が絶えず維持されるようなプロセスを構築し、そのプロセスが妥当であることなどが評価されることになります。
IDATEN制度は今後の活用が期待されますが、当面の間、PMDAの医療機器開発前相談を申し込み、変更計画がIDATEN制度の対象になるかどうかについて事前に助言を受けることになっています。
(2021年6月30日時点の記載です。)
(注1)先駆け審査指定制度とは、世界に先駆けて開発され早期の治験段階で著明な有効性が見込まれる医薬品等を指定し、優先審査等の対象とする仕組み(小児の用法用量設定といった特定用途医薬品等)
(注2)条件付き早期承認制度とは、患者数が少ない等により治験に長期間を要する医薬品等を、一定の有効性・安全性を前提に、条件付きで早期に承認する仕組み
(注3)薬生機審発0831第14号「医療機器の変更計画の確認申請の取扱いについて」(令和2年8月31日)
(注4)事務連絡「医療機器の変更計画の確認申請に関する質疑応答集(Q&A)について」(令和2年10月30日)
臨床研究法は2018年4月1日に施行され、厚生労働省より臨床研究を実施する上での疑問点に対するQ&Aや事例集などが発出注1)されています。しかし、多様な医療機器では解釈に困難が伴うことが多いのも事実です。
「臨床研究法の対象となる臨床研究」は、①「医薬品・医療機器等を人に対して投与または使用する研究」、②「医薬品・医療機器等の有効性又は安全性を明らかにすることを目的とした研究」の2つ要件を共に満たすものとなっています。さらに、「人に対して投与又は使用する行為のうち、医行為に該当するもの」と規定されています。
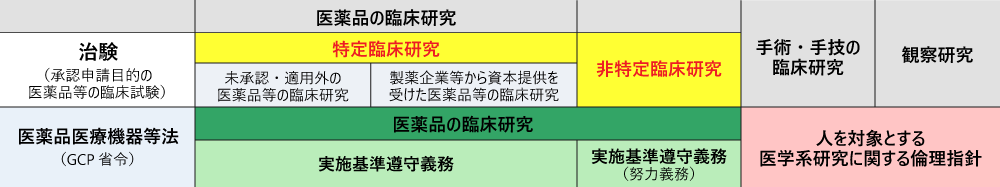
(厚生労働省ホームページの資料より一部改変)
「臨床研究法の対象となる臨床研究」には、「特定臨床研究」と「非特定臨床研究」があります。このうち、(a)未承認・適応外の医薬品等を⽤いて実施する臨床研究、(b)製薬企業等から資⾦提供を受けて実施する臨床研究が「特定臨床研究」となります。 このため、未承認での機器開発の段階では、まず、「法の対象となる臨床研究」の該当性を判断することが必要です。その判断におけるポイントは以下の通りです。
| 臨床研究法の該当性―判断ポイント | 非該当の事例 |
| 1) 医療機器に該当しないものか? | 福祉機器等 |
| 2) 薬機法に基づくものか? | 治験、再審査・再評価等の製造販売後調査、適合性試験等 |
| 3) 医療機器の評価を伴わないものか? | 手術・手技の評価、バイオマーカ探索・病態解明、使用感の評価等 |
| 4) 人に対して投与・使用しないものか? | 生体試料を用いた検査機器の性能評価等 |
| 5) 医行為を伴わないものか? | 侵襲性のない検査機器の性能評価等 |
| 6) 観察研究か? | 患者のために最も適正な医療を提供した結果として診療情報又は試料の収集により得られた情報を利用する研究 |
(厚生労働省講演資料より一部改変)
このうち、「医行為」は「医師の医学的判断及び技術を持ってするのでなければ人体に危害を及ぼし、又は及ぼす恐れのある行為」とされていますが、具体的に個別に判断する必要があるため、その範囲は明確ではなく、特に、低リスクの新しい診断機器を開発している医工学の研究者にとって大きな問題となっています。このため、日本生体医工学会では、「医工学研究における臨床研究法の該当性判断ガイドライン」注2)を作成し、公開しています。
(2019年12月19日時点の記載です。)
(注1)厚生労働省ホームページ「臨床研究法について」
(注2)日本生体医工学会ホームページ「臨床研究法の該当性判断ガイドライン」
2014年11月25日に医薬品医療機器等法(薬機法;「医薬品、医療機器等の品質、有効性及び安全性の確保等に関する法律」)が改正施行され、ソフトウェア単体も「医療機器プログラム(SaMD:Software as a Medical Device)」として医薬品医療機器等法の規制の対象になりました。
「医療機器プログラム」(注1)は、①疾病の診断、治療、予防に寄与するなど、医療機器としての目的性を有し、かつ、②意図したとおりに機能しない場合に患者(又は使用者)の生命及び健康に影響を与えるおそれがあるプログラム(ソフトウェア機能)となっており、汎用PCやモバイル端末等にインストールして医療機器としての機能を与えたり、これまでの医療機器と組み合わせたりして使用されます。
このため、同じ機能を有するプログラムであっても、使用目的によって医療機器の該当性の判断が異なります。また、クラスⅠ(機能の障害等が生じた場合でも人の生命及び健康に影響を与えるおそれがほとんどないもの)に相当するプログラムは医療機器の範囲から除外されていることが、これまでの医療機器とは大きく異なっています。
「医療機器プログラム」は、①疾病診断用プログラム、②疾病治療用プログラム、③疾病予防用プログラムの3つに類別されています。当初の医療機器プログラムは、既承認の医療機器に搭載されていたソフトウェアを、プログラム単体として認証・承認を受けたものがほとんどでしたが、最近では、AI技術を活用した画像診断支援などの革新的な医療機器プログラムが次々と承認されています。また、行動変容を促す治療アプリが承認されるなど、モバイル端末などを医療やヘルスケアに利用するデジタル医療、デジタルヘルスとしても注目されています。
こうした「プログラム医療機器」の開発や実用化を加速するために、様々な取り組みが進められています。例えば、2020年9月には、プログラム医療機器のバージョンアップに速やかに対応できる制度として、「医療機器の特性に応じた変更計画の事前確認制度(IDATEN制度)」(Q8参照)がスタートし、同年11月には、厚生労働省から「プログラム等の最先端医療機器の審査抜本改革(DASH for SaMD)」が公表され、最先端のプログラム医療機器の早期実用化を促進するための一元的窓口として「医療機器プログラム総合相談」がPMDAに設置されています。
また、「医療機器プログラム」の該当性に関しては、「プログラムの医療機器への該当性に関する基本的な考え方について」(2014年11月14日)が発出されていましたが、これをより明確化・精緻化するために、「プログラムの医療機器該当性に関するガイドラインについて」(2021年3月31日)(注2)が公表されました。これには、基本的な考え方に加えて、該当性判断事例も記載されています。
さらに、2022年6月7日に閣議決定された「規制改革実施計画」の「プログラム医療機器(SaMD)に関する承認審査等の見直し」に基づき、「医療機器プログラム(SaMD)の審査ポイント」や「医療機器プログラムの認証基準」の公表・策定(注3)が進められています。前者は、2014年以降に承認を取得した「医療機器プログラム」から、審査のポイントに関する情報を整理して順次公表するもので、後者は、承認実績が存在する医療機器プログラムについては、認証基準の策定及び改正を行うものとなっています。また、2022年度の診療報酬改定では、プログラム医療機器の新たな診療報酬上の評価についての検討が進み、「プログラム医療機器等医学管理加算」の項目が新設されています。
こうした変化の激しい分野では、研究開発や臨床研究を実施する上で、PMDAのホームページなどで常に最新の動向や取り組みについて把握しておくことが重要となります。
(注1)「医療機器プログラム」はプログラム単体として流通するもので、プログラムを記録した記録媒体も含めたものは「プログラム医療機器」と呼ばれています。
(注2)薬生機審発0331第1号・薬生監麻発0331第15号「プログラムの医療機器該当性に関するガイドラインについて」(令和3年3月31日)
(注3)PMDA「医療機器プログラム(SaMD)の審査ポイント」(参照2022-12-16)
(2022年12月16日時点の記載です。)
医療機器プログラムにおいても、有効性の評価のために、人を対象として承認申請のための試験を実施する場合、原則、GCPに準拠した臨床試験(治験)になります。
一方で、例えば、画像診断プログラムの有効性は、非典型的なものを含めて様々な症例画像に対して評価することが求められます。しかし、治験で求められている前向きでの症例画像の収集方法では、患者同意を取得しても、それが非典型的な症例の画像であるかどうかは撮像後でないと分かりません。このため、評価対象の画像を効率的に収集することは困難で、現実的でないことも多々あります。
こうした場合、治験ではなく、体外診断機器の有効性評価で用いられている臨床性能試験の枠組みを利用することが可能でした。この取り組みは、当初、例外的位置づけとなっていましたが、令和3年9月29日に発出された通知「追加的な侵襲・介入を伴わない既存の医用画像データ等を用いた診断用医療機器の性能評価試験の取扱いについて(薬生機審発0929第1号)」(注1)において、「性能評価試験」として、以下の通り明確化されました。
この通知は、人工知能技術を利用した医用画像診断支援システムやDNAシークエンサーを利用した遺伝子変異解析システム等の診断用医療機器を対象とし、追加的な侵襲・介入(診断結果の伝達を含む。)を伴うことなく、「既存の医用画像データ又は生体試料及びこれらに関連する既存の診療情報等」(医用画像データ等)を収集して実施する性能評価試験が対象となっています。この医用画像データ等には、通常の診療で得られたものだけでなく、バイオバンク、データベース等において提供されているものも含まれますが、介入を伴う臨床研究等で得られるデータは除外されています。
「性能評価試験」は、診療情報を使用するかどうかで、以下の2種類に分けられます。
- 既存の医用画像データ又は生体試料のみを収集し、新たに評価上必要な情報等を付ける等した上で、性能評価に用いる場合
これは、試験に使用するデータ等の信頼性確保のために、原資料(カルテ情報等)との照合ができるようにしておく必要がない場合で、承認申請時の添付資料の位置付けは、「設計及び開発に関する資料」になります。 - 既存の医用画像データ又は生体試料及びこれらに関連する既存の診療情報を収集し、性能評価に用いる場合
これは、試験に使用する診療情報の信頼性確保のために、原資料(カルテ情報等)との照合ができるようにしておく必要がある場合で、承認申請時の添付資料の位置付けは、「臨床試験の試験成績に代替する資料」になります。
後者の場合、例えば、評価対象の画像の正解データとして確定診断の情報等用いることが可能となりますが、「試験に使用するデータ等の第三者への提供・開示及び承認申請を含む商用利用に関する患者等の同意が適切に得られていることについて、承認申請時にPMDAの求めに応じて申請者が根拠資料に基づいて説明できること。」となっていることに留意する必要があります。
(注1)「追加的な侵襲・介入を伴わない既存の医用画像データ等を用いた診断用医療機器の性能評価試験の取扱いについて(令和3年9月29日薬生機審発0929第1号)」
(2022年10月1日時点の記載です。)
特定機能病院注1)における重大な医療事故の発生を踏まえて、医療安全対策の強化の方向性が検討された結果、特定機能病院の承認要件の見直しが行われました。
特定機能病院においては、診療科の長は、次の①と②については、あらかじめ、特定機能病院の管理者が設置する部門(「担当部門」)に申出を行い、承認を得る体制に変更されます。
①「高難度新規医療技術」を用いた医療の提供
②「未承認新規医薬品等」を用いた医療の提供
ここでの「高難度新規医療技術」とは、当該病院で実施したことのない医療技術(軽微な術式の変更等を除く。)であって、その実施により患者の死亡その他の重大な影響が想定されるものになります。例えば、既承認の医療機器であっても、患者の死亡その他の重大な影響が想定されるもので、当該病院で実施したことのない医療技術に当たる場合は、①の対象になります。また、「未承認新規医薬品等」とは、当該病院で使用したことのない医薬品又は高度管理医療機器であって、医薬品、医療機器等の品質、有効性及び安全性の確保等に関する法律(薬機法)における承認又は認証を受けていないものになります。つまり、未承認医療機器を用いた臨床研究で、当該病院でこれまで使用したことがないリスクの高い高度管理医療機器(クラスⅢ、Ⅳ)を用いた場合は、②の対象になります。
各特定機能病院は、厚生労働省が発出した基準注2),3)に従って規程を作成注4)し、臨床研究を実施する診療科等はこの規程を遵守することになります。具体的には、対象となる臨床研究は、新設の「高難度新規医療技術評価委員会」、あるいは「未承認新規医薬品等委員会」と倫理審査委員会の両方で審査を受けることになります(既存の倫理審査委員会が上記の基準を満たす場合には、評価委員会を兼ねることもできます)。なお、臨床研究法に基づき認定臨床研究審査委員会で審査される臨床研究については、「高難度新規医療技術評価委員会」、あるいは「未承認新規医薬品等委員会」の審査は不要です。提供する医療が、「高難度新規医療技術」や「未承認新規医薬品等」に該当するか否かは、一義的には診療科の長の判断によりますが、判断が困難な場合には「担当部門」の意見を聞くことになります。また、病院で実施したことのある医療技術、使用したことのある医療機器であっても、従来の実施体制に大きな変更があった場合には、診療科の長は、改めて適切な実施体制の確認を行う必要があります。
これらの特定機能病院の承認要件の変更は、2016年6月10日付けで交付され、2017年3月には経過措置が終了したため、2017年4月から運用が開始されています。また、一般的な病院においても、高度な医療技術を提供する場合などには、特定機能病院に対する規定を参考に、同様の取り組みを求める努力義務が課されることになります。
(2020年1月24日時点の記載です。)
(注1)平成5年4月施行の医療法の第2次改正によって制度化された医療機関の機能別区分のうちのひとつ。高度な医療を提供する医療機関について、厚生労働大臣が個別に承認するもので、承認要件が定められている。
(注2)医政発0610第21号「医療法施行規則第9条の23 第1項第7号ロの規定に基づき高難度新規医療技術について厚生労働大臣が定める基準について」(平成28年6月10日)
(注3)医政発0610第24号「医療法施行規則第9条の23 第1項第8号ロの規定に基づき未承認新規医薬品等を用いた医療について厚生労働大臣が定める基準について」(平成28年6月10日)
(注4)「高難度新規医療技術の導入プロセスに関するQ&A」(高難度新規医療技術の導入プロセスにかかる診療ガイドライン等の評価・向上に関する研究班作成)
医療機器のユニークデバイス識別(Unique Device Identification: UDI)は、医療機器の製品仕様の日本米国欧州間の統合整合化を審議する国際医療機器規制当局者フォーラム(IMDRF)が2011年に発行した「医療機器ユニークデバイス識別ガイダンス」に挙げられている①個々の医療機器に対するバーコード識別表示と②国家レベルの製品情報のデータベース登録に対応する措置のことを指します。①医療機器の識別表示では、i)製品機器識別情報として、製品名、製品コード(機種)、製造業者名などの固定データとともに、ii)製造識別情報として、機器の有効期限日、ロット番号、シリアル番号などの可変(=機器毎に固定でない)データを示すことが求められています。また、②データベース登録については、各国の保健当局によって対応が異なります。
UDIの運用は、医療機器や医療材料に関して、それぞれの製品情報を中央管理し、トレーサビリティーを確保することにより、医療安全の向上(医療機器に関連した合併症や医療過誤の原因解明と予防策の構築、感染対策、医療機器使用の標準化・平均化)を主たる目的とし、医療機器流通に掛かる負担軽減(リコール・返品業務の効率化、偽造医療機器の排除)と行政視点での医療機器に掛かる医療費の不適切な保険償還請求の防止もその目的になっています。
UDIは、米国FDAでは2014年から医療機器クラスに応じて罰則規定のある法律として義務化されていますが、欧州委員会では検討段階で未施行です。日本(厚生労働省)ではバーコードによる医療機器の識別表示が罰則規定のない省令として求められていますが、データベースによる中央管理は実施されていません。したがって、国内で開発製品化された医療機器を米国に向けて輸出する際にはUDI対応を検討しなければいけないことになります。ただし、IMDRFのガイダンスに基づくUDI対応は、日本としても国際整合性の確保から避けられず、近い将来に厚生労働省から通知が発出され、米国向け輸出機器だけでなく、国内向けおよび欧州向け医療機器でもUDI対応が義務化されることが予想されます。米国FDAのデータベース(Global UDI Database: GUDID)はweb上で公開されており、誰でも医療機器を検索し、その情報をダウンロードできるようになっています。例えば、MRI対応の可否や再滅菌の可否などの情報を得ることが出来ます。
それでは、医療機器の臨床研究を行う際に、UDI対応はどのような意味を持つでしょうか?国内で臨床研究の対象となる適応外・未承認医療機器は、医療機器識別バーコード表示の義務化ルールの範囲外でありUDI対応は必須ではありません。ただし、UDIの理念にある医療安全の観点は、臨床研究における副作用発現時の原因究明や臨床研究の継続にとって重要な評価となりますので、UDIを意識した医療機器管理は試験を実施する研究者にとって有用であると考えられます。
(2020年1月24日時点の記載です。)
(参考文献)
黒澤康雄. FDA, 各国規制当局のUDI規制の進捗 –規制をクリアした医療機器の市場対応と輸出拡大のために-. 医療機器学 2015: 85: 345-53
医療機器の承認等においては、「品質」、「有効性」及び「安全性」が評価の対象となります。一方、特許の出願では、「産業上利用可能性」、「新規性」、「進歩性」が要件となり、加えて、特許を受けようとする発明が明細書に明確に記載され(「明確性の要件」)、実施可能なものであって(「実施可能要件」)、発明の詳細な説明に記載した範囲を超えるものであってはならない(「サポート要件」)とされています(注1)。
医薬品の場合は、一般に物の構造や名称からその物をどのように作り、どのように使用するかを理解することが比較的困難な技術分野に属する発明であることから、医薬発明(注2)として、記載要件などについて特許審査の運用を明確化した審査基準が作成されています(注3)。
この医薬発明では、有効成分となる化合物等の薬理作用(新たに発見した属性)により、当該化合物等が特定の疾患の治療(新たな用途)に適することを裏付ける資料が、発明の詳細な説明に記載されていることが必要とされています。このため、有効成分となる化合物等の薬理作用、特定の疾患の治療に適することを裏付ける資料として、通常、有効成分の化合物等に関する薬理試験結果の記載が必要となります。この薬理試験として、臨床試験、動物実験あるいは試験管内実験が挙げられています。発明の詳細な説明の記載が不足していた場合、出願後に試験成績を提出してそれを補うことは出来ないため注意する必要があります。
一方、医療機器の場合は、他の産業分野の発明と同様に、その構造や動作原理から機能や効果を示すことが比較的容易であることが多く、薬理試験のように有効性を示すデータが求められることは余りありません。このため、試作品で評価を行う前のアイデア段階でも特許出願が可能となっています。ただし、特定の用途など、技術常識や文献等からだけでは、発明の詳細な説明が不足していると判断される場合は、医薬品と同様に試験成績等の裏付けを求められることに留意する必要があります。特に、近年、デジタルヘルスの出現により医薬品と医療機器の境界が判り難くなっています。プログラム医療機器においては、AI機能などを搭載することでその物の用途と効果との一体性が明確でなくなっています。こうした場合、発明の効果を主張する上で有効性を示す試験成績等のデータは効果的です。
また、米国では日本では認められていない「人間を手術、治療又は診断する方法の発明」などの医療関連行為発明が特許権として認められていることについても留意する必要があります。日本で、「物の特許」として出願した後、国際出願をして米国で「方法の特許」としての特許権取得を目指す場合、有効性を示すデータを当初の明細書に盛り込んでおくとより広い範囲で権利化できる可能性があります。
(2022年10月1日時点の記載です。)
(注1)「特許・実用新案 審査ハンドブック(特許庁)」
(注2)医薬発明は、ある物の未知の属性の発見に基づき、当該物の新たな医薬用途を提供しようとする「物の発明」であり、「医薬用途」とは、以下の(i)又は(ii)を意味します。
(i)特定の疾病への適用
(ii)投与時間・投与手順・投与量・投与部位等の用法又は用量が特定された、特定の疾病への適用
(注3)「特許・実用新案 審査ハンドブック 附属書B 第3章 医薬発明(特許庁)」
国内の薬機法注1)で規制される医療機器は「人若しくは動物の疾病の診断、治療若しくは予防に使用されること、又は人若しくは動物の身体の構造若しくは機能に影響を及ぼすこと」を目的としており、「介護」は含まれていません。これに対して、介護機器や福祉用具は、福祉用具法注2)で「心身の機能が低下し日常生活を営むのに支障のある老人又は心身障害者の日常生活上の便宜を図るための用具及びこれらの者の機能訓練のための用具並びに補装具」と規定されており、薬機法の規制対象外になっています。 一方、EUでは、医療機器規則(Medical Device Regulation; MDR)において、「疾患の診断、予防、モニタリング、効果予測、予後予測、治療又は緩和」に加えて「傷害又は障害(disability)の診断、モニタリング、治療、緩和又は補償(compensation)」が医療機器の使用目的に含まれています。このため、介護機器や福祉用具も医療機器として規制対象になります。また、米国FDAでも、例えば、車椅子は、理学療法機器(Physical Medicine Devices)の中の理学療法用補装具(Physical Medicine Prosthetic Devices)として位置づけられ、医療機器として規制されています。一般的な車椅子は、EUでは手動、電動ともにクラスⅠの医療機器であり、米国、中国などでは、手動車椅子はクラスⅠ、電動車椅子はクラスⅡの医療機器となっています。
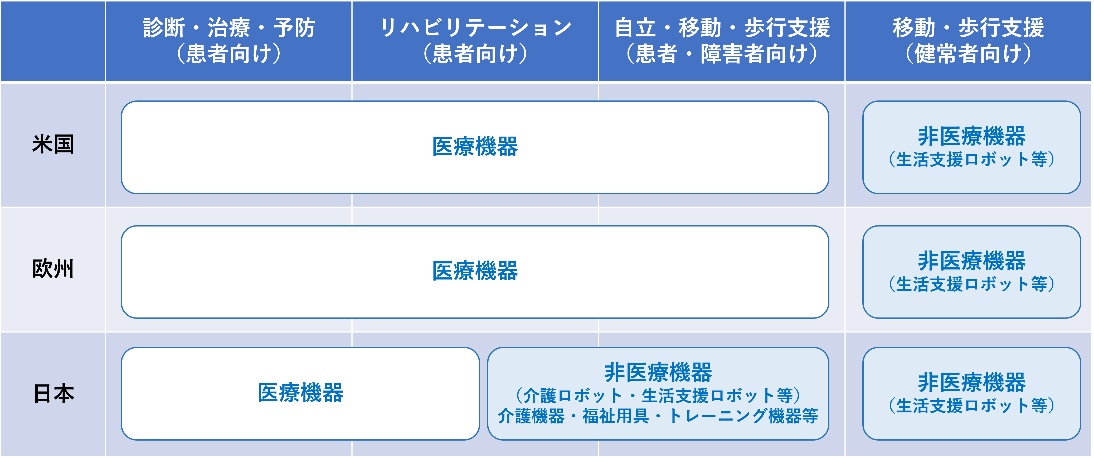
世界的に高齢化が進む中、利用者の自立支援や介護者の負担軽減に繋がる生活支援ロボットや介護ロボットの開発が期待されています。国内では非医療機器であったとしても、これらを海外に展開する場合、図に示すように、医療機器として規制の対象になることに十分留意する必要があります。
(2024年3月1日時点の記載です。)
(注1)薬機法:医療機器等の品質、有効性及び安全性の確保等に関する法律
(注2)福祉用具法:福祉用具の研究開発及び普及の促進に関する法律
国内の「臨床評価報告書」は、本来は臨床試験(治験)が必要な品目に対して、既存の文献等を用いて臨床上の有効性及び安全性を評価できる場合に作成するもので、臨床試験成績(治験成績)に代えて提出する承認申請のための添付資料になります。一方、EUの「臨床評価報告書(Clinical Evaluation Report ; CER)」は、医療機器のライフサイクル全体にわたって行われる臨床評価注1)の結果を文書化したもので、全てのクラス分類の医療機器注2)において実施が求められます。このため、国内の「臨床評価報告書」はEUと比較して狭義となっています。
EUの臨床評価は、使用説明書に基づいて機器を使用した場合に、医療機器規則(Medical Device Regulation; MDR)の「安全性と性能に関する一般的要求事項」(General Safety and Performance Requirements; GSPR)に機器が適合していること、リスク/ベネフィットが受容可能であることを、臨床データによる十分な臨床的エビデンスに基づいて示すことが目的となります。このため、製造業者は、当該機器や同等機器に関して保有している臨床データ、文献検索から得られる臨床データ、また、当該分野の最新知識や最先端技術に関する情報を収集し、それを評価、分析して、臨床評価報告書を作成する必要があります。この「臨床評価報告書」は機器の技術文書の一部と位置付けられています。
機器によっては、文献検索による臨床データが、臨床エビデンスの大部分を占める場合があります。このため、文献検索と文献レビューは慎重に計画され、プロトコルを作成して包括的かつ批判的なアプローチで行うことが求められます。一方で、既存の臨床データや文献だけではエビデンスが不十分な場合は、臨床試験(clinical investigation)注3)を実施することで、新たに臨床データを取得する必要があります。臨床試験の必要性は、機器の使用目的やリスク/ベネフィットなどを考慮して判断する必要があります。高リスクのクラスⅢや埋込み型の医療機器では、原則、臨床試験が要求されます。
(2024年3月1日時点の記載です。)
(注1)EUの医療機器指令(Medical Devices Directive93/42/EEC; MDD)では、臨床評価のガイダンスMEDDEV 2.7/1 rev.4 “Clinical evaluation: Guide for manufacturers and notified bodies”(June 2016)が発行されていましたが、医療機器規則(Medical Device Regulation 2017/745; MDR)では、それに代わるガイダンスがいまだ発行されていません。このため、ガイダンスが発行されるまでは、引き続きMEDDEVに従う必要があります。ただし、MDRとの間に相違がある場合、MDRが優先されます。
(注2)EUでは、医療機器はクラスⅠ、クラスⅡa、クラスⅡb、クラスⅢにクラス分類されています。
(注3)EUでは、”clinical investigation”の用語が、clinical trial (臨床試験)またclinical study (臨床研究)と同義として使用されています。
薬機法注1)の第2条第14項において、「体外診断用医薬品とは、専ら疾病の診断に使用されることが目的とされている医薬品のうち、人又は動物の身体に直接使用されることのないものをいう。」と規定され、国内では、汎用検査用試薬のグルコースキットなど、体外診断IVD(In vitro diagnostics)は試薬としての名称が使用されています。しかし、体外診断薬は、その特性から、薬機法において、「医療機器及び体外診断用医薬品」や「医療機器又は体外診断用医薬品」と医療機器と並記されることが多く、実質、医療機器と同様の取り扱いになっています。なお、体外診断用医薬品の製造販売承認申請注2)などで測定機器を使用する場合は、使用機器は一般的な名称(分光光度計、血液自動分析装置、血球計数器等)や専用装置の名称を用い、その操作方法は試薬側から見た標準的な手順を記載すること注2)になっています。
一方、FDAは、体外診断用製品を「疾病またはその後遺症を治癒、緩和、治療、予防するために、健康状態の判定を含む疾病またはその他の状態の診断に使用することを意図した試薬、器具、システムで、人体から採取した検体の収集、調製、検査に使用することを意図したもの」と定義しており、これらは連邦食品医薬品化粧品法(Federal Food, Drug, and Cosmetic Act; FD&C Act)で規定される機器として扱われます。(公衆衛生法(Public Health Service Act)の対象となる生物学的製剤である場合もあります。)
また、EUの体外診断用医療機器規則(In Vitro Diagnostic Regulation; IVDR)においても、体外診断用医療機器(in vitro diagnostic medical device)を「製品が体外で使用されることを製造者が意図している、または主としてそれが用いられることを意図している、人体から採取された血液や組織などの検体の検査のため試薬、試薬製品、検量物質、管理物質、キット、器具、装置、ソフトウェア、またはシステムで、単独で使用されるか、または組み合わせて使用されるかを問わない」と規定し、試薬を含めて医療機器として位置付けています。
これらは、国際医療機器規制当局フォーラム(International Medical Device Regulators Forum; IMDRF)注3)が、「診断、モニタリング、適合性の目的のために情報提供することを単独または主目的として、人体由来の検体を体外で検査することを製造業者により意図された、単独または組み合わせて使用される機器であり、これには、試薬、検量物質、管理物質、試料採取容器、ソフトウェア、および関連する機器・装置またはその他の物品が含まれる。」とする定義と一致しています。
(2024年3月1日時点の記載です。)
(注1)薬機法:医療機器等の品質、有効性及び安全性の確保等に関する法律
(注2)薬食機参発1121第16号「体外診断用医薬品の製造販売承認申請に際し留意すべき事項について」 (平成26年11月21日)
(注3)IMDRFは、国際的な医療機器規制の調和と収束を加速させることを目的とした医療機器規制当局の任意団体